Tykk- og endetarmskreft (tarmkreft) er for både for menn og kvinner den nest hyppigste kreftformen i Norge (1). I 2007 ble 3 375 pasienter diagnostisert med tarmkreft, hvorav ca. 2/3 var lokalisert i tykktarmen og resten i endetarmen (1). Insidensen av tarmkreft i Norge har vært kraftig økende de siste 50 år (2), men det er ting som tyder på at den dramatiske økningen er i ferd med å flate ut. For tykktarmskreft er kjønnsfordelingen lik, mens flere menn enn kvinner (1,5:1) får endetarmskreft (1), uten at man med sikkerhet vet årsaken til dette. Selv om median alder ved debut er om lag 70 år, opptrer tarmkreft i alle aldersgrupper, også hos helt unge. Hos disse foreligger ofte predisponerende faktorer som inflammatorisk tarmsykdom eller arv (3). Andre risikofaktorer for utvikling av tarmkreft er trolig røyking, alkohol, stort inntak av bearbeidet rødt kjøtt, lite inntak av fiber og inaktivitet (2).
Materiale og metode
Artikkelen er basert på klinisk erfaring med behandling av tarmkreft og anbefalinger fra Norsk Gastrointestinal Cancer Gruppe. I databasen PubMed er det brukt søkeordene ”colorectal cancer” og “MSI, CEA, hereditary, targeted treatment”. Videre har vi brukt kunnskap ervervet gjennom eget forskningsprosjekt om tarmkreft og arv.
Arvelig tarmkreft
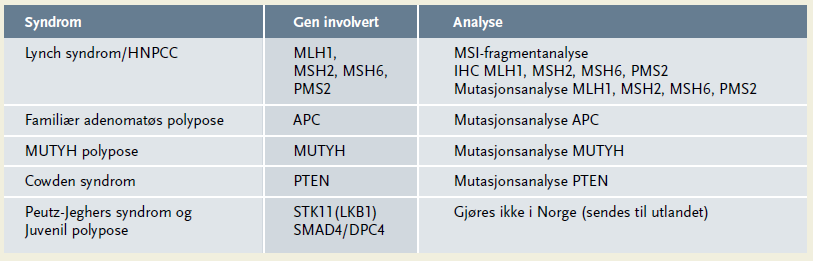
Det er beregnet at 25 - 30 prosent av pasienter med tarmkreft har en familiehistorie med tarmkreft, inklusive 5 - 10 prosent som har et arvelig syndrom forårsaket av mutasjoner i ett enkelt gen (4). Den vanligste formen for arvelig tarmkreft, Lynch syndrom (også kalt Hereditær ikke-polypøs kolorektal cancer; HNPCC), er årsak i 3 - 5 prosent av tilfellene. Lynch syndrom er forårsaket av feil i ett av fire DNA mismatch reparasjonsenzymer (MLH1, MSH2, MSH6 eller PMS2). Familiær polypose er involvert i cirka én prosent. De hyppigste årsakene er feil i enten APC-genet (Familiær adenomatøs polypose; FAP) eller MUTYH-genet (MUTYH-polypose). Oversikt over de kjente arvelige tarmkreftsyndromene finnes i tabell 1 (3).
Hos 15 - 25 prosent sees en familiær opphopning uten at den bakenforliggende genetiske årsaken er kjent. I disse tilfellene er det trolig en mer sammensatt årsak av både lavpenetrante genvarianter og miljøfaktorer.
Diagnostikk
Symptomer på tarmkreft er ofte endring i avføringsvaner (diaré/obstipasjon), blod eller slim i avføringen og eventuelt jernmangelanemi (5). Symptomer på tarmobstruksjon kan også foreligge. Ved langtkommen sykdom kan symptomer som nedsatt matlyst, vekttap og redusert allmenntilstand opptre. Det er svært viktig at blod i avføringen hos en tidligere frisk person tas på alvor, og at det fører til henvisning til ano/rekto/kolonoskopi. Det skjer ofte at diagnostiseringen blir forsinket fordi pasienten feilaktig får hemoroider som diagnose. Dersom pasienten selv har observert blod er det unødvendig å sjekke for occult blod i avføringen med for eksempel Hemofec®/Hemoccult-II®. Dette fordi svulster kan blø periodevis; en negativ test for occult gastrointestinal blødning utelukker derfor hverken blødning eller svulst.
Diagnosen stilles ved rekto/kolonoskopi med biopsi (1). CT thorax/abdomen/bekken hører med i preoperativ kartlegging for å avklare spredningsstatus. I tillegg utføres MR bekken dersom det foreligger cancer i rektum (6). Dette for å vurdere om svulsten lar seg operere primært, eller om det er behov for preoperativ strålebehandling kombinert med cellegift (radiokjemoterapi).
Patologi
De aller fleste svulster i tarm er adenokarsinomer. Kun i få prosent av tilfellene foreligger sjeldne typer som nevroendokrin svulst, lymfom, plateepitelkarsinom eller andre. En patologisk beskrivelse skal inneholde opplysninger om grad av differensiering (lav, middels, høy), grad av mucinøst og signetringcelleinnslag, hvorvidt det foreligger karinvasjon og lymfocyttinfiltrasjon, og hvorvidt det er spredning til lokale/regionale lymfeknuter. For en fullstendig undersøkelse med hensyn på prognostisering, kreves det at minst 12 lymfeknuter er undersøkt (5).
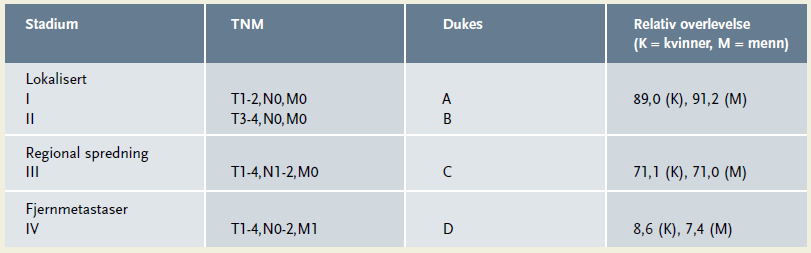
TNM stadieinndelingen som brukes for å beskrive svulstenes utbredelse ved diagnosetidspunktet, beskriver tumorens størrelse (T), hvorvidt det foreligger metastaser til lokale/regionale lymfeknuter (N), og om det foreligger fjernmetastaser (M) (7) (tabell 2). TNM klassifikasjonen har tatt over etter Dukes klassifikasjonen, men Dukes brukes ofte fortsatt parallelt med TNM (tabell 3).

Blodprøver
Det tas blodprøver rutinemessig for å sjekke hematologisk status, lever- og nyrefunksjon (5). Tumormarkøren karsinoembryonalt antigen (CEA) tas rutinemessig når kreft i tarmen er påvist. CEA er ikke egnet som screeningmarkør for tarmkreft, men brukes i oppfølgingen av pasienten for å påvise et eventuelt tidlig residiv eller som hjelp i monitorering av behandlingseffekt (5, 8). I det siste er det reist tvil om nytten av CEA i den postoperative oppfølgingen av pasienter med tarmkreft (9). CEA kan være forhøyet også ved andre kreftformer, og i enkelte tilfeller også hos personer uten kreft (inflammatorisk tarmsykdom som ulcerøs colitt og Crohns sykdom, divertikulose i tykktarm, leverlidelser og lungeinfeksjoner). Røykere har normalt litt høye CEA-verdier. Ulike immunologiske metoder brukes for å måle CEA i blod, og dette utføres på medisinsk biokjemisk laboratorium.
Prognose
Overlevelsen ved tarmkreft er betydelig bedret siden 1950-tallet (1). Overlevelsen er sterkt avhengig av stadiet ved diagnosetidspunktet, og TNM-stadiet er fortsatt den viktigste prognostiske faktor ved tarmkreft. Dette til tross for at det de siste 10-15 år er utført storskala molekylærbiologisk kartlegging av tarmkreft, og kunnskapen om tarmkrefts utvikling, genotype og fenotype har økt kolossalt. Helbredelsesprosenten er høy (90 prosent) ved stadium II uten lymfeknutemetastaser, mens den er omkring 70 prosent dersom lymfeknutemetastaser foreligger (tabell 3) (10). Om lag halvparten av pasientene utvikler metastaser i forløpet av sykdommen, og om lag 25 prosent har metastaser ved diagnosetidspunktet (5, 10). Relativt få av disse helbredes, men median overlevelse har økt betraktelig det siste tiåret etter introduksjon av mer effektive cellegifter (20 måneder) (5, 11).
Behandling
De fleste pasienter opereres primært med fjerning av modersvulsten (5, 11). Hvis det foreligger fjernmetastaser på diagnosetidspunktet (om lag 25 prosent), vurderes behandlingsstrategien ut fra flere faktorer og helst etter drøfting i tverrfaglige team (onkolog, kirurg, radiolog og eventuelt patolog). I dag er det mer vanlig at pasienter som har metastaser på diagnosetidspunktet (synkrone metastaser) starter med kjemoterapi i stedet for å bli operert først, men kirurgi av primærsvulsten vurderes selvsagt i de tilfeller der obstruksjon truer pasienten. I slike tilfeller kan en avlastende stomi eller stent være et alternativ til kirurgi.
Ved lokalavansert endetarmskreft gis det preoperativ radiokjemoterapi i fem uker (en 2 Gys fraksjon daglig mandag-fredag), og pasienten opereres seks - åtte uker etter avsluttet behandling (5, 12). Det er vurdert at om lag 30-40 prosent av pasienter med endetarmskreft har behov for slik neoadjuvant behandling.
Hvis histopatologisk undersøkelse viser at det foreligger spreding til lymfeknuter (N1-2; Dukes C), skal pasienten tilbys postoperativ (adjuvant) cellegift i 6 måneder (5, 10). En slik behandling er vist å bedre overlevelsen med omlag 12 prosent (5, 10).
Hvis pasienten har fjernspredning, skilles mellom mulig kurativ, livsforlengende palliativ og symptomlindrende palliativ behandling. Siste år har andelen pasienter som opereres for lever- og lungemetastaser vært økende, og flere studier viser at fem års overlevelse ved metastasekirurgi ligger rundt 30 – 40 prosent (5, 13). Slik kirurgi kombineres ofte med cellegiftbehandling.
Flere cellegifter benyttes i behandlingen av tarmkreft. 5-fluorouracil (5-FU) har vært brukt i over 50 år, og er fremdeles hjørnesteinen i behandlingen. På slutten av 90-tallet ble det vist at både irinotekan og oksaliplatin er effektive cellegifter ved denne typen kreft, og i dag brukes ulike kombinasjoner av disse tre cellegiftene (5, 11).
Målstyrt terapi og KRAS
Storskala molekylærbiologisk kartlegging av ulike kreftsvulster har ført til en eksplosiv utvikling av kreftmedisiner som er rettet mot spesifikke mål på/i kreftcellen eller dens nærmiljø. Slik målstyrt/skreddersydd terapi er også tatt i bruk ved tarmkreft.
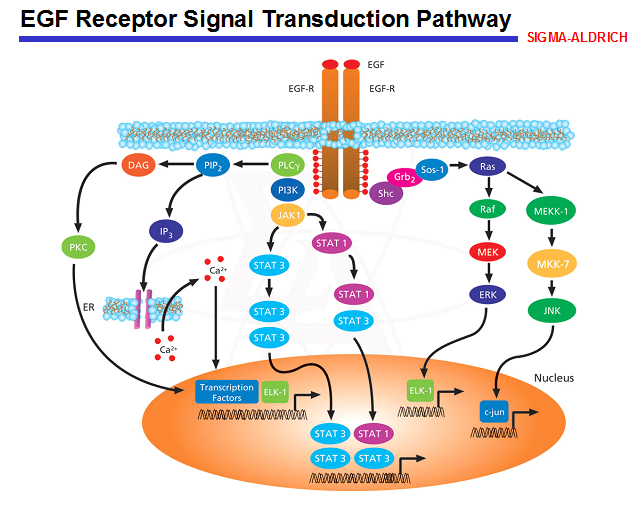
Det har lenge vært kjent at epidermal vekstfaktor-reseptoren (EGF-R) er overuttrykt i majoriteten av tarmkreft. Antistoff rettet mot EGF-R eller småmolekylære stoffer som går intracellulært og blokkerer ulike trinn i EGF-R signalveien (tyrosinkinasehemmere), er utviklet og testet ut i en rekke studier. Ved tarmkreft er det først og fremst antistoff som har vist seg å være effektive, og antistoffene cetuximab og panitumumab er i bruk i 3. linjes behandling ved metastatisk tarmkreft (5, 11). I 2008 kom det et viktig gjennombrudd i forskningen omkring bruken av antistoff mot EGF-R ved tarmkreft, da det ble vist at kun pasienter med vill-type av KRAS-genet har nytte av slik behandling (5). Ingen pasienter med mutert KRAS har nytte av antistoff rettet mot EGF-R (cetuximab og panitumumab). Mutasjonen i KRAS er aktiverende, og de muterte cellene står og fyrer uavhengig av hva som skjer på overflaten (figur 1). De mest aktuelle stedene i KRAS-genet kan testes for forekomst av mutasjon i DNA isolert fra tumorvev (primærtumor eller metastaser). I tumorvevet vil det alltid være en viss andel normalceller tilstede, og det er viktig ved utvelgelse av prøvemateriale at det er høy forekomst av tumorceller, helst mer enn 50 prosent.
Bevacizumab er et antistoff som er godkjent brukt i 1. linjes behandling av metastatisk tarmkreft, og da i kombinasjon med 5-FU/irinotekan (5, 11). Antistoffet er rettet mot VEGF (vaskulær endotelial vekstfaktor), og griper inn i neovaskulariseringen av kreftsvulsten. På grunn av høye kostnader ved bruk av antistoff mot EGF-R og VEGF, har helsemyndighetene i Norge besluttet at bruken av disse skal registreres i såkalt fase IV studier (studier som gjøres etter at det er gitt markedsføringstillatelse for et medikament) (14).
Tarmkreft og arv
Å identifisere individer med en økt arvelig risiko for tarmkreft er viktig for å kunne gi disse et kontrollopplegg som kan redusere dødeligheten forårsaket av denne sykdommen. Kontrollopplegget består av jevnlig koloskopi, og ved funn av forstadier til kreft (adenomer), blir disse fjernet. For å finne familiens genfeil, må man ha en blodprøve fra en affisert slektning. Hvis det blir identifisert en genfeil hos en person som har vært syk, og denne genfeilen gir økt risiko for sykdom, kan denne benyttes som en prediktiv/presymptomatisk test hos slektninger.
Når det gjelder den molekylærgenetiske mekanismen for tarmkreft, skilles det mellom den mikrosatelitt instabile (MSI) veien (cirka 15 prosent) og den kromosomal instabile veien (cirka 85 prosent) (figur 2) (15).
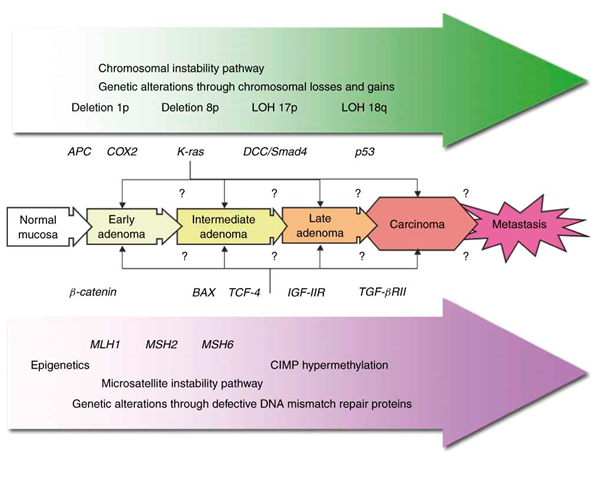
Den mikrosatelitt instabile veien kan igjen inndeles i sporadisk tarmkreft (skyldes hypermetylering av MLH1 promotoren) og arvelig Lynch syndrom (kimbanemutasjon i MLH1, MSH2, MSH6 eller PMS2). MSI oppstår fordi replikasjonsfeil ikke blir reparert når det foreligger feil i DNA mismatch reparasjons (MMR) systemet. Dette kan sees som endret lengde av mikrosatelittsekvenser (1-2 repeterte nukleotider) i DNA fra tumorvev, sammenlignet med normalvev. Som normalvev kan blodprøve benyttes. For å få en indikasjon på hvilket gen som er defekt, kan det gjøres immunohistokjemi med antistoffer mot MLH1, MSH2, MSH6 og PMS2. Hvis det er bortfall av protein, er det en indikasjon på at det er feil i dette genet. For å finne genfeilen, blir aktuelt gen sekvensert.
Hvis det er bortfall av MLH1-protein, kan dette skyldes metylering av MLH1-promotoren. Derfor kan det gjøres metyleringsanalyse av MLH1-promotoren for å skille mellom arvelig og sporadisk MSI tarmkreft. En annen indikasjon på at det er en sporadisk MSI tarmkreft, er forekomst av en spesiell mutasjon i onkogenet BRAF (i kodon 600), som ikke forekommer ved Lynch syndrom.
Gendiagnostikk for de arvelige polyposesyndromene gjøres ved å direkte sekvensere APC- og MUTYH-genene. Tabell 1 viser en oversikt over genanalysene som gjøres i Norge for diagnostikk av arvelig tarmkreft.
Oppsummering
Tarmkreft rammer over 3000 nordmenn årlig, og skyldes trolig både arv og ulike miljøfaktorer. Funn av blod i avføringen skal tas på alvor og føre til utredning. Ved tidlig debut må arvelige kreftsyndromer eller familiær predisposisjon mistenkes og utredes. Prognosen avhenger av sykdomsutbredelsen ved diagnosetidspunktet. Behandlingen er multimodal, og tett samarbeid mellom ulike spesialister er nødvendig for å vurdere den riktige behandlingen for hver enkelt pasient. Målstyrte kreftmedisiner er tatt i bruk i behandlingen av disse pasientene. Både overlevelsen totalt, samt leveutsikter ved metastatisk sykdom, er bedret de siste årene.