FAG resymé
Hemoglobinopati på avveie – α-talassemi hos pasienter av norsk opprinnelse
α-Talassemi forekommer hyppig i de tropiske og subtropiske områdene av verden og er svært sjeldent hos personer av skandinavisk opprinnelse. En studie utført ved Avdeling for medisinsk biokjemi ved Oslo universitetssykehus viste fire sjeldne former for α-talassemi hos 20 pasienter av norsk opprinnelse.
Av RUNA MARIE GRIMHOLT
Sykdommens årsak og konsekvenser
α-Talassemi skyldes hovedsakelig store delesjoner som strekker seg over ett eller begge α-globingenene på kromosom 16 (HBA2 og HBA1). Sekvensvarianter og delesjoner av det regulatoriske området HS-40 er mer sjeldent. Normalt har man fire α-globingener, to på hvert kromosom. Grovt sett kan man si at alvorlighetsgraden av α-talassemi følger antall gener som er mutert. Den mest alvorlige formen, hvor alle fire α-globingenene er deletert, kalles Hb Barts hydrops føtalis og er i de aller fleste tilfellene ikke forenlig med liv. Bærertilstander av α-talassemi er derfor viktig å påvise for å kunne gi tilstrekkelig genetisk veiledning til bærere som ønsker barn.
Om studien
I denne studien ble 20 pasienter inkludert på bakgrunn av talassemisk fenotype og norsk opprinnelse. Alle prøvene ble undersøkt med standard hemoglobinopatiutredning i perioden 2012 – 2018. Dette innebærer hematologi, jernstatus, hemoglobintyping og α-talassemi gentest (påviser syv vanlige delesjoner som gir α-talassemi). Resultatene fra disse undersøkelsene viste at samtlige pasienter hadde mikrocytose, normal jernstatus og negativ α-talassemi gentest. For å påvise andre mutasjoner som årsak til pasientenes fenotype, ble α-globingenene undersøkt med DNA-sekvensering for å påvise sekvensvarianter, og kvantitativ PCR med TaqMan-prober (HBA-CNV-analyse) for å påvise store delesjoner av α-globingenene og HS-40.
Funn
DNA-sekvensering av α-globingenene påviste en enkeltbase-delesjon (HBA2:c.345del) som ble funnet hos åtte pasienter fra seks ulike familier. Sekvensvarianten var tidligere rapportert i databasene ItheGenes, dbSNP og ClinVar, og var klassifisert som patogen. Hos en pasient påviste sekvenseringen en delesjon av 20 baser (HBA2:c.142_161del), som ikke tidligere var beskrevet i litteraturen. Begge disse delesjonene førte til et skift i leserammen og et prematurt stoppkodon. Siden introduksjon av tidlig stoppkodon er en kjent molekylær mekanisme for α-talassemi, kunne vi med dette forklare pasientenes hematologiske forandringer.
En stor delesjon av begge α-globingenene ble detektert ved hjelp av HBA-CNV-analyse hos åtte pasienter fra tre forskjellige familier. Karakterisering av bruddpunktene viste at delesjonen var 13,4 Kb stor (figur 1) og ikke tidligere publisert.
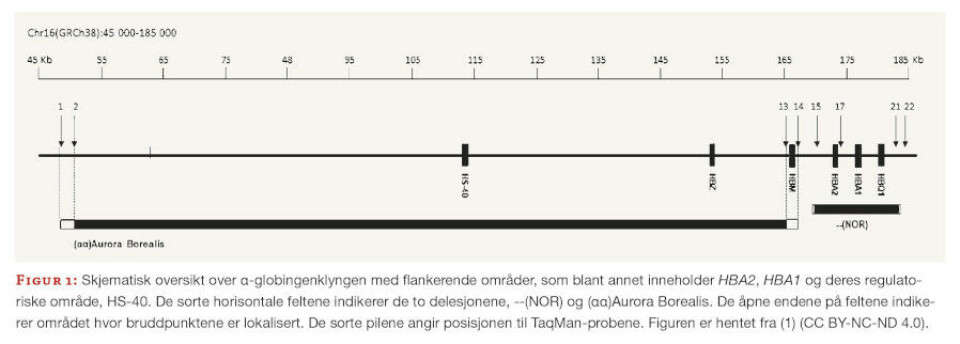
Delesjonen fikk navnet delNOR (--(NOR)) og kunne forklare pasientenes vedvarende mikrocytose. HBA-CNV ble også brukt til å påvise en delesjon av HS-40-regionen (figur 1) hos tre pasienter fra samme familie. HS-40 er essensiell for ekspresjon av α-globingenene, og flere delesjoner i dette området er kjent for å gi α-talassemi. Delesjonen var cirka 120 Kb stor og ble kalt (αα)Aurora Borealis.
Konklusjon
De hematologiske forandringene ved talassemi kan lett forveksles med jernmangel, spesielt hos pasienter av skandinavisk opprinnelse hvor talassemi er svært sjeldent. Flere av pasientene i denne studien hadde blitt feilaktig behandlet med jerntilskudd uten effekt. Resultatene fra denne studien viser at muligheten for talassemi hos pasienter av norsk opprinnelse ikke må ignoreres, selv om det er svært sjeldent.
























