Fag i praksis
Komplementsystemets rolle i aterosklerose
Komplementsystemet har en nøkkelrolle i kolesterolkrystallindusert inflammasjon iaterosklerose. Studier fra NTNU, Senter for molekylær inflammasjonsforskning (CEMIR), harvist hvordan kolesterolkrystaller gir en inflammatorisk respons i arterieveggen.
Komplementsystemets rolle i aterosklerose.pdf(187 KB)
Av NATHALIE NIYONZIMA
Aterosklerose og dannelse av kolesterolkrystaller
Hjerte- og karsykdommer forårsaker globalt 17,9 millioner dødsfall per år, og var med dette den ledende dødsårsak i verden i 2019 (1, 2). De vanligste formene er koronar hjertesykdom (angina pectoris og hjerteinfarkt) og cerebrovaskulær sykdom (hjernedrypp, hjerneslag og hjerneblødning). Den underliggende årsaken for disse tilstandene er aterosklerose, en lidelse som utvikler seg tregt og resulterer i dannelsen av en kronisk betennelse i åreveggen. Over tid kan dette bli livstruende.
Fra å være identifisert som en feil ved kolesterollagring, er nå aterosklerose anerkjent som en sykdom som skyldes en kronisk betennelse (3). Kunnskapen om bidraget fra immunforsvaret i aterosklerose, fra dannelse av aterosklerotiske plakk til destabiliseringen av eksisterende plakk som fører til blodpropp, er stadig økende. Men hva som starter betennelsen i åreveggen er imidlertid ikke godt beskrevet. Kolesterol som akkumuleres i aterosklerotiske plakk, i form av kolesterolester i skumceller eller som utfelte
kolesterolkrystaller (CC), kan starte utvikling og progresjon av sykdommen (4).
Man finner CC i alle fasene av aterosklerose, og krystallene setter fart i betennelsen man ser i utviklingen av sykdommen. Kroppen identifiserer CC som skadelige stoffer. Våre forsvarsceller, kalt makrofager, klarer ikke å bryte ned og rydde vekk disse CC. I stedet setter makrofagene i gang en betennelse når de prøver å fjerne CC.
CC i aterosklerose
CC er en viktig bidragsyter tidlig i aterogenesen som gir en kraftig immunrespons. Via aktivering av det intracellulære proteinkomplekset kalt inflammasom (NLRP3) blir interleukin-1β (IL-1β) frigjort (4) (se faktaboks med ordforklaringer). IL-1β er et potent cytokin som driver immunresponsen videre. CC er også til stede i plakk hos mennesker (5-7).
Selektivhemming av IL-1β for å forhindre koronare hendelser har allerede vært i klinisk utprøvning. Resultater etter behandling med medikamentet Canakinumab (humant monoklonalt antistoff rettet mot IL-1β) viste reduserte betennelsesreaksjoner hos pasienter med hjerteinfarkt, og det reduserte også tilbakevendende kardiovaskulære hendelser, sammenlignet med placebo (8).
Komplementsystemet
Komplementsystemet er en viktig del av det medfødte immunsystemet som umiddelbart registrerer fremmede inntrengere. Hovedoppgaven er en rask gjenkjenning av fremmede strukturer, som for eksempel mikrober eller strukturer som eksponeres i skadet vev (9). I tillegg vil komplementsystemet gjenkjenne strukturer som dannes ved såkalt steril inflammasjon, altså inflammasjon som er forårsaket av krystaller ved utfelling av kolesterol i aterosklerose eller av urinsyre i leddgikt. Ved bakterielle infeksjoner bidrar komplementsystemet blant annet til opsonisering, kjemotakse og bakteriedrap. I normal tilstand er komplementsystemet kroppens forsvarsvenn, men det kan skade organer og eventuelt bidra til en kronisk tilstand som aterosklerose når det kommer ut av kontroll.
Komplementproteiner har tradisjonelt blitt definert som plasmaproteiner. De siste årene har en rekke oppdagelser vist at komplementsystemet er ett av immunsystemets viktigste forsvarsverk. Det finnes både på utsiden og inne i immunceller (10-13). Forskning viser at funksjonen til de ulike komplementproteinene defineres av deres lokalisasjon. For eksempel vil komplementproteiner i plasma fungere forskjellig, sammenlignet med de samme proteinene på innsida av celler.
Ved CEMIR har vi sett nærmere på mekanismene som ligger bak CC-indusert betennelse i aterosklerose. I de neste delkapitlene drøfter jeg arbeid som er gjort på CEMIR for å studere komplementsystemets bidrag i CC-indusert betennelse, arbeid som har økt kunnskap om CC-betennelsesreaksjoner i aterosklerose.
Ekstracellulært komplement fasiliterer fagocytose av CC
I en studie vi gjorde i 2014, så vi at ekstracellulære komplementproteiner var viktige i CC-indusert betennelse i aterosklerose (14). Vi benyttet oss av en human fullblodsmodell, der alle blodets bestanddeler var til stede og kunne påvirke hverandre. Blodet ble inkubert med CC, og vi fant ut at CC aktiverer komplementsystemet og dermed klargjør inflammasomet NLRP3 for aktivering. Videre så vi at komplementsystemet er viktig for opsonisering og fagocytose av CC. I denne studien brukte vi blod fra en pasient med en sjelden genetisk defekt der komplement 5 (C5) mangler helt (15). Denne delen av komplementsystemet er viktig for å danne sluttproduktet C5b-9, som fører til bakteriedrap. Sammenlignet med blod fra friske, kunne vi kartlegge at C5 var viktig i den inflammatoriske responsen til CC. I tillegg brukte vi hemmere av deler av komplementsystemet, som førte til at opptak av CC – og dermed produksjonen av en rekke cytokiner - blir redusert, inkludert IL-1β (14).
Celleproduserte komplementproteiner
Betydning i plakk
Celler i plakk produserer og uttrykker komplementproteiner, både på mRNA- og proteinnivå (16). Komplementproteiner er uttrykt i celler fra både stabile og ustabile plakk. Ved CEMIR har vi nylig vist at noen av disse proteinene lokaliseres rundt CC i plakk, særlig i områder med nekrose (17). I dette arbeidet benyttet vi oss av perifere mononukleære blodceller (PBMC) og karotisplakk fra pasienter med forskjellige grader av aterosklerose. Vi studerte betennelse ved hjelp av forskjellige analysemetoder, inkludert ELISA, immunhistokjemi, konfokal mikroskopi og helgenomsekvensering.
Vi fant at komplementproteiner var sterkt uttrykt i plasma fra pasienter med både stabile og ustabile plakk, sammenlignet med plasma fra friske personer. Dette tyder på økt systemisk betennelse i begge pasientgruppene. I karotisplakk akkumulerte komplement C1q og C5b-9 rundt CC, og det var økt genuttrykk av komplementreseptorene; C3aR, C5aR1 og C5aR2 fra karotisplakk fra pasienter med akutt koronart syndrom. Dette tyder på at lokalt komplement (produsert av cellene) i plakk er driver av aterosklerose. Det kan tenkes at bindingen av komplement på CC i plakk kan fungere som kjemotakse som tiltrekker flere immunceller til CC i plakk, og dermed vekst av plakk. Dette arbeidet demonstrerte en positiv korrelasjon mellom CC- aktivering av komplementsystemet i aterosklerotisk plakk med alvorlighetsgrad av aterosklerose (17).
Betydning på innsiden av celler
I arbeidet vårt som ble publisert i Science Immunology (18) benyttet vi oss av isolerte makrofager fra blod fra friske frivillige givere, samt musemodeller og plakk fra pasienter, for å studere betennelse i aterosklerose. Når vi tilførte CC til makrofager, så vi at intracellulært komplement C5a og reseptor C5aR1 ble aktivert, og at disse proteinene kolokaliserte i makrofager (Figur 1).
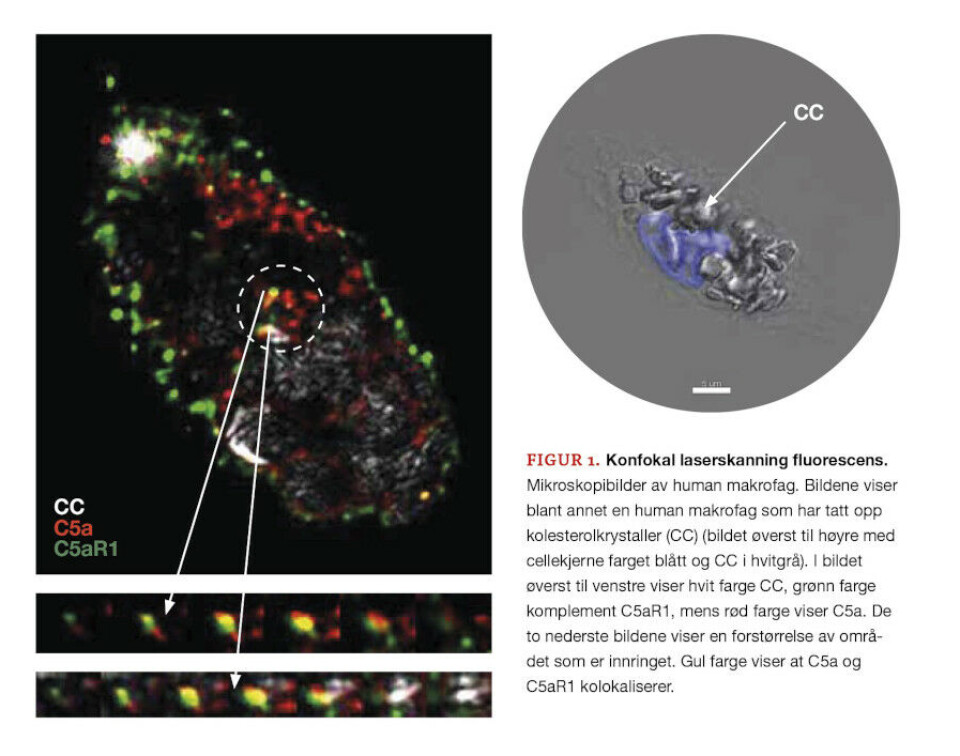
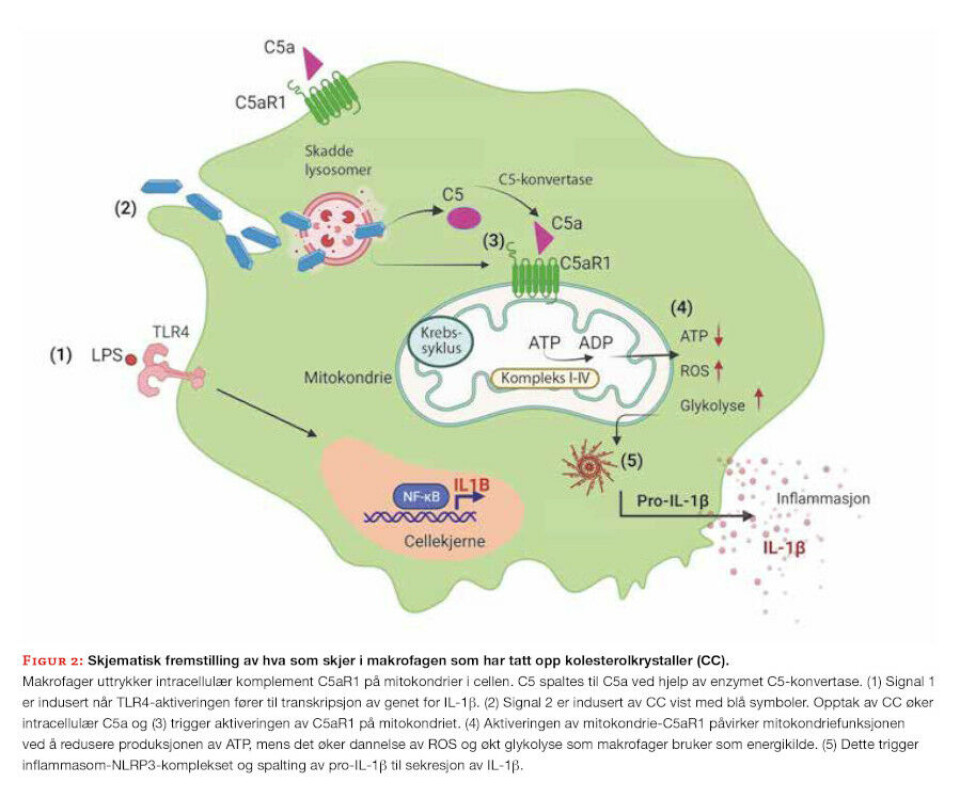
Vi så at dette utløste en betennelsesreaksjon ved at makrofagene startet å lage det potente cytokinet IL-1β, som er vist å akselerere utviklingen av aterosklerose. Ved å hemme det intracellulære komplementsystemet kunne betennelsesreaksjonen i makrofagene dempes. I dette arbeidet oppdaget vi at komplementreseptoren C5aR1 sitter på mitokondriene.
Når makrofagene tok opp CC ble komplementreseptoren på mitokondriene aktivert og utløste produksjon av betennelsesfremmende reaktive oksygenforbindelser (ROS), samt økning i ATP og glykolyse for å forsyne makrofagene med energi. Våre funn tyder på at C5aR1 på mitokondriene fungerer som et alarmsystem som oppdager CC, og setter i gang produksjon av IL-1β og betennelse i aterosklerose.
For å bekrefte vårt funn etablerte vi en musemodell for aterosklerose, der genene for C5aR1 eller C5 var modifisert kun i makrofager, men ikke i andre immunceller. Vi fant ut at fjerning av C5aR1-genet reduserte størrelse og grad av betennelse i aterosklerotiske plakk. Vi tilførte også hemmere av intracellulær C5aR1 til plakk fra pasienter med aterosklerose, og dette reduserte den generelle betennelsen i plakkene betydelig. I samme studie fant vi at genetisk modifisert C5 eller C5aR1 i makrofager også beskyttet mus mot nyresvikt. Denne studien viser at det intracellulære komplementsystemet er en viktig faktor i den biologiske prosessen som fører til steril betennelse som oppstår i aterosklerose (Figur 2). Vårt arbeid peker også på muligheten av å benytte hemmere av intracellulært komplement for behandling av aterosklerose.
Konklusjon
Studier har vist at systemisk og intracellulært komplementaktivering står sentralt i CC-indusert betennelse i aterosklerose. Mens ekstracellulært komplement er viktig for fagocytose av CC med en gang makrofager har tatt opp CC, så er det intracellulært komplement C5aR1 på mitokondriene som oppdager CC og setter i gang en kaskade som utløser produksjon av potente cytokin IL-1β. Resultatene er viktige for å forstå mekanismer bak aterogenese, og understreker betydningen av å hemme intracellulært komplement C5a-C5aR1-akse i steril inflammasjon.
























